CRISPRi干扰中心代谢基因转录对苏氨酸合成的影响
刘旭峰1,王宁1,郝亚男1,李英滋1,范晓光1,2,谢希贤1,2*
1(代谢控制发酵技术国家地方联合工程实验室(天津科技大学),天津,300457) 2(天津市微生物代谢与发酵过程控制技术工程中心,天津,300457)
摘 要苏氨酸是重要的饲料氨基酸,需求量持续增加,提高苏氨酸发酵产率和糖酸转化率,降低生产成本已成为一个重要课题。该实验以苏氨酸工程菌Escherichia coli THRD为出发菌,利用CRISPRi(clustered regularly interspaced short palindromic repeats interference)技术研究中心代谢9个基因转录水平的改变对苏氨酸合成的影响。发酵结果显示,干扰zwf、pfkA和gltA基因的转录水平提高了苏氨酸的合成效率,对应菌株苏氨酸产量分别为60.3、64.6和65.8 g/L,与出发菌(50.9 g/L)相比,分别提高了18.5%、26.9%和29.3%。糖酸转化率分别为40%、38%和39%,与出发菌(34%)相比,分别提高了17.7%、11.8%和14.7%。结果表明,通过CRISPRi干扰中心代谢基因的转录水平,可以调节合成代谢网络,使更多碳源流向苏氨酸,提高苏氨酸的合成效率。同时,该研究也为其他生物制品工程菌的构建提供了参考。
关键词大肠杆菌;苏氨酸;代谢干扰;糖酸转化率;CRISPRi
苏氨酸化学名为α-氨基-β-羟基丁酸,是人体必需的8种氨基酸之一[1]。主要应用于医药卫生、食品强化剂、饲料添加剂等方面。苏氨酸是继谷氨酸和赖氨酸,世界第三大氨基酸产品[2]。大肠杆菌由于繁殖迅速、发酵温度高、生理生化基础研究比较深入等特征,成为苏氨酸生产最主要的菌株。苏氨酸工程菌的构建经历了2个重要的阶段。(1)诱变育种阶段,日本味之素株式会社椎尾勇等,经诱变育种筛选出有产苏氨酸能力的菌株;(2)基因工程阶段,研究人员运用代谢工程技术与系统生物学方法构建苏氨酸工程菌,如韩国LEE等[3]。
苏氨酸作为大宗氨基酸产品,提高发酵产率和糖酸转化率可显著降低生产成本,提高经济效益。常规基因工程技术和代谢工程技术一般采用加强主要代谢途径,阻断竞争代谢途径以减少代谢分流来提高产品合成效率,但是一些竞争途径的代谢基因敲除经常会对细胞生长造成负面影响[4-5]。代谢干扰可以根据需要调节基因转录和蛋白表达水平,进而优化代谢网络,提高合成效率[6]。目前,代谢干扰主要方法有反义RNA技术和CRISPRi技术[7-8]。反义RNA技术因具有操作简单,适用范围广泛,特异性强以及安全性高的特点被应用于代谢干扰研究。反义RNA技术中sRNAs(small regulatory RNAs)调控基因转录技术应用较为广泛。sRNAs由2个部分组成:支架序列和目标绑定序列,在大肠杆菌中自然形成二级结构,该二级结构能够识别目标mRNA并为Hfq蛋白提供1个支架,使目标mRNA降解或三者结合形成的复合物阻碍核糖体与目标mRNA的结合,从而调节相关基因的转录水平。NA等[9]以1,5-戊二胺工程菌E.coli XQ56为出发菌,利用sRNAs技术调控基因murE表达,在高密度培养的条件下,对应菌株产酸较原菌XQ56高31%。CRISPRi技术是基于RNA引导的DNA核酸内切酶的Ⅱ型CRISPR/Cas9系统发展而来,与CRISPR/Cas9系统不同之处在于前者的Cas9蛋白失去了DNA核酸内切酶的活力[10]。CRISPRi干扰体系由dCas9蛋白和sgRNA两个组件构成,其中与dCas9蛋白共同表达的sgRNA由3部分组成:20 nt目标基因特定核苷酸区域的识别序列、42 nt dCas9蛋白结合序列以及40 nt转录终止子序列[11-13]。CRISPRi工作机制为当成熟的sgRNA引导dCas9蛋白结合在DNA的特定位置时,三者形成的复合物能够影响转录延伸、RNA聚合酶以及转录因子的结合,从而实现调节基因表达水平的目的。CRISPRi技术具有灵活高效的特点,可以调控任意基因转录水平,该技术现在也已经被广泛应用于代谢工程领域和合成生物领域[14-21]。
本研究以苏氨酸工程菌E.coli THRD为出发菌,依据CRISPRi干扰机制[18, 20],建立了基于阿拉伯糖诱导的CRISPRi代谢干扰系统,该系统首先将dcas9基因整合在基因组上并受控于阿拉伯糖启动子,同时,用pGRB质粒表达特异的sgRNA。利用上述干扰体系研究中心代谢途径主要基因的转录水平的改变对苏氨酸合成的影响。发酵结果表明,调节zwf、pfkA以及gltA基因的表达水平可以提高工程菌的苏氨酸合成效率。
1 材料与方法
1.1 菌株与质粒
苏氨酸工程菌E.coli THRD保藏在天津科技大学菌种保藏中心,实验所涉及的菌种及质粒见表1。
表1 本实验所用菌株及质粒
Table 1 Strains and plasmids in this study

菌株/质粒特征来源E. coli DH5αthe cloning host本实验室E. coli THRDthreonine producer (ILEL, AHVr)本实验室THRD-1ILEL, AHVr, mlc::mlc∗, araBAD::dcas9本研究pRedCas9Cas9 expression vector, Sper[22]pGRBsgRNA expression vector, Ampr[22]pGRB-aceE1plasmid carrying aceE sgRNA targeting ATG region of aceE gene本研究pGRB-aceE2plasmid carrying aceE sgRNA targeting the central region of aceE gene本研究pGRB-aceE3plasmid carrying aceE sgRNA targeting the tail region of aceE gene本研究pGRB-gltA1plasmid carrying gltA sgRNA targeting ATG region of gltA gene本研究pGRB-gltA2plasmid carrying gltA sgRNA targeting the central region of gltA gene本研究pGRB-pck1plasmid carrying pck sgRNA targeting ATG region of pck gene本研究pGRB-pck2plasmid carrying pck sgRNA targeting the central region of pck gene本研究pGRB-pfkA1plasmid carrying pfkA sgRNA targeting ATG region of pfkA gene本研究pGRB-pfkA2plasmid carrying pfkA sgRNA targeting the central region of pfkA gene本研究pGRB-pykF1plasmid carrying pykF sgRNA targeting ATG region of pykF gene本研究pGRB-pykF2plasmid carrying pykF sgRNA targeting the central region of pykF gene本研究pGRB-sucA1plasmid carrying sucA sgRNA targeting ATG region of sucA gene本研究pGRB-sucA2plasmid carrying sucA sgRNA targeting the central region of sucA gene本研究pGRB-sucC1plasmid carrying sucC sgRNA targeting ATG region of sucC gene本研究pGRB-sucC2plasmid carrying sucC sgRNA targeting the central region of sucC gene本研究pGRB-sucD1plasmid carrying sucD sgRNA targeting ATG region of sucD gene本研究pGRB-sucD2plasmid carrying sucD sgRNA targeting the central region of sucD gene本研究pGRB-zwf1plasmid carrying zwf sgRNA targeting ATG region of zwf gene本研究pGRB-zwf2plasmid carrying zwf sgRNA targeting the central region of zwf gene本研究
1.2 培养条件与培养基
大肠杆菌THRD工程菌及完成基因编辑的菌株培养条件为LB培养基、37 ℃培养,含有pRedCas9质粒的菌株培养条件为LB培养基、32 ℃培养;大肠杆菌DH5α用于构建和克隆质粒,培养条件为LB培养基、37 ℃培养。氨苄青霉素工作质量浓度为100 μg/L;奇霉素工作质量浓度为100 μg/L。
LB培养基,2-YT培养基和SOC培养基参见分子克隆实验指南。
苏氨酸种子培养基配方:25 g/L蔗糖、10 g/L酵母粉、6 g/L蛋白胨、1.2 g/L KH2PO4、0.5 g/L MgSO4、 10 mg/L MnSO4、10 mg/L FeSO4、1.3 mg/L VB1、0.3 mg/L VH。
苏氨酸发酵培养基:30 g/L葡萄糖、2 g/L酵母粉、4 g/L蛋白胨、1 g/L柠檬酸钠、2 g/L KH2PO4、0.7 g/L MgSO4、0.1 g/L MnSO4、0.1 g/L FeSO4、0.8 mg/L VB1、0.2 mg/L VH。
1.3 CRISPR/Cas9基因编辑方法
利用CRISPR/Cas9技术对菌株进行基因编辑[22-24]。CRISPR/Cas9系统包括pGRB和pRedCas9两个质粒,其中pRedCas9质粒包含pGRB的消除系统、重组酶、Cas9蛋白表达系统和奇霉素抗性;pGRB质粒包括gRNA序列、Cas9蛋白结合区域序列、终止子序列和氨苄青霉素抗性。构建质粒pGRB的目的是为了转录相应sgRNA,与Cas9蛋白形成复合体并识别靶位点,使Cas9蛋白准确切割双链目的基因。同时,在重组酶的作用下,通过同源重组将目的DNA片段整合在基因组上,完成基因编辑。
1.3.1 构建gRNA质粒和DNA片段的获得
为了构建gRNA质粒,先设计合成2条反向互补的单链DNA,通过退火形成双链DNA,该双链DNA中间序列为靶位点的特定gRNA间隔序列,两端序列与pGRB具有同源序列。双链DNA与线性化pGRB通过同源重组形成gRNA表达质粒。
利用引物设计软件primer 5,以待编辑基因的上下游序列为模板,设计上下游同源臂引物;以待整合基因为模板,设计整合基因的扩增引物。通过PCR的方法分别扩增上下游同源臂和目的基因片段,再经过重叠PCR制备重组片段。
1.3.2 菌株构建
首先,将pRedCas9质粒通过电转导入E.coli THRD感受态细胞中。通过菌落PCR筛选正确的阳性转化子,并将该菌株接种于LB培养基32 ℃过夜培养,然后按1%的接种量转移到100 mL 2-YT培养基中,32 ℃ 培养细胞OD600达到0.1~0.2,添加0.1 mmol/L IPTG诱导重组酶表达,继续培养细胞OD600达到0.4~ 0.5,收集菌体制备电转感受态细胞。将200 ng重叠DNA片段和100 ng gRNA质粒混匀在电转感受态细胞中,并转移至0.1 cm电转杯中,用Eppendorf电转仪在1 850 kV条件下将质粒和DNA片段导入细胞,同时加入1 mL SOC中32 ℃复苏2 h,然后取100 μL 涂布到含氨苄和奇霉素抗性的LB平板上,32 ℃ 过夜培养。随机挑取单菌落进行菌落PCR验证,根据菌落PCR的DNA片段大小确定阳性重组菌株。将阳性菌株在含有0.3 g/L阿拉伯糖的LB培养基中培养,诱导pRedCas9中pGRB消除系统将pGRB消除;由于pRedCas9是温敏型质粒,通过将菌株在42 ℃下培养消除pRedCas9质粒,获得无质粒工程菌株。
1.4 干扰质粒及菌株构建
干扰质粒与gRNA质粒构建方法相同,方法同上。将构建成功的干扰质粒电转至以THRD-1为基础的苏氨酸大肠杆菌工程菌中,取100 μL稀释涂布到含氨苄抗性的LB平板上,37 ℃过夜培养,筛选阳性菌株。
1.5 苏氨酸发酵
将菌株经斜面活化后接种于装液量为30 mL的种子培养基中(500 mL摇瓶),37 ℃,200 r/min条件下培养10 h。然后以10%接种量转接到发酵培养基中,并在发酵培养4 h添加诱导剂阿拉伯糖,终质量浓度控制在0.6 g/L。在37 ℃,200 r/min条件下发酵培养24 h。
1.6 发酵过程中检测与分析
大肠杆菌生物量的测定利用分光光度计在600 nm处的吸光度(OD600)检测。
发酵液中残余的葡萄糖及合成的有机酸采用高效液相分析仪(HPLC)检测。检测方法:取1 mL发酵液,13 000 r/min离心2 min留取上清。将上清液过膜处理后,开始进行色谱分离。色谱分离条件:色谱柱为AminexRRHPX-87H(300 mm×7.8 mm),流动相为5 mmol/L H2SO4,柱温为30 ℃,检测波长为215 nm,流动相总流速为0.5 mL/min,采用等浓度洗脱的方法。
苏氨酸采用高效液相分析仪(HPLC)柱前衍生测定。衍生方法:取1 mL发酵液,13 000 r/min离心2 min留取上清。在1.5 mL离心管中加入200 μL衍生缓冲液(42%(质量分数)NaHCO3溶液),加入10 μL 发酵液混匀后,再加入衍生试剂溶液(1%(质量分数)2,4-二硝基氟苯乙腈溶液)300 μL摇匀,将离心管置于65 ℃水浴锅中反应1 h后取出。将样品冷却至室温后,加入定容缓冲液(68%(质量分数)KH2PO4溶液)690 μL摇匀,然后将样品过膜处理,开始进行色谱分离。色谱分离条件:色谱柱为Agilent ZORBAX Eclipse AAA(4.6 mm × 150 mm,5 mm),流动相为乙腈-乙酸钠缓冲液,柱温为33 ℃,检测波长为360 nm,流动相总流速为1 mL/min, 采用二元梯度洗脱的方法。
1.7 总RNA的提取和实时荧光定量PCR
根据aceE、dcas9和E.coli W3110 16S rDNA(rrnb,内参)基因序列采用Premier 5.0 软件设计用于RT-PCR的引物。发酵培养10 h取样,并参照Promega试剂盒说明书要求提取RNA,参照RR036A试剂盒进行反转录,RT-PCR体系及方法参照SYBRR Premix EX-Taq TM Ⅱ试剂盒说明书。首先扩增目的基因和内参基因,通过分析扩增曲线指数增长期的平行性来确定扩增倍数作为后续实验的标准,然后对菌株的目的基因和内参基因进行定量测定(每个样本3个重复),并用ΔΔCt法分别计算基因aceE和dcas9相对转录量。ΔΔCt法计算流程:步骤一,内参基因均一化样品差异,即Ct(目的基因)-Ct(内参基因)= ΔCt;步骤二,实验样品和对照样品比较,即ΔCt(实验样品)- ΔCt(对照样品)= ΔΔCt;步骤三,使用公式计算,即倍数变化=2-ΔΔCt,式中Ct(循环阈值)即PCR扩增过程中扩增产物的荧光信号达到设定的荧光阀值时所经过的扩增循环数。
1.8 发酵数据统计学分析
发酵数据代表3组平行发酵数据的平均值和标准偏差。利用T检验双尾分布对2组发酵参数进行单向方差分析。0.01<P< 0.05显示显著,用“*”表示;P< 0.01显示极其显著,用“**”表示。
2 结果与分析
2.1 阿拉伯糖诱导代谢干扰系统的构建
目前,有关CRISPRi干扰技术的研究均采用dcas9基因和sgRNA共表达体系,该体系通过单质粒表达实现调节基因转录水平的目的。但由于dcas9基因过大,采用单质粒过表达的方式,会增加菌体生长负担。为了减轻干扰系统引入对细胞生长的影响,本实验首先构建了一套基于阿拉伯糖诱导的干扰体系。利用CRISPR/Cas9基因编辑方法,以苏氨酸工程菌E.coli THRD为出发菌,将dcas9基因整合在基因组的araB、araA、araD基因位置位点,受控于阿拉伯糖启动子Para。在代谢工程研究中,通过诱导调控基因表达是常用的改造策略,常用的诱导剂有乳糖、木糖、四环素和阿拉伯糖等。比较而言,阿拉伯糖诱导具有严紧性以及对菌体没有毒害作用的特点[25]。随后为了减少葡萄糖对阿拉伯糖启动子的阻遏,根据文献报道[26],对mlc基因启动子-10区进行点突变,解除葡萄糖效应,构建了菌株THRD-1。最后,将干扰特定基因的sgRNA连接到质粒pGRB上,通过电转导入THRD-1菌株中,构建相应的干扰菌株。
2.2 阿拉伯糖诱导条件下的代谢干扰体系测试
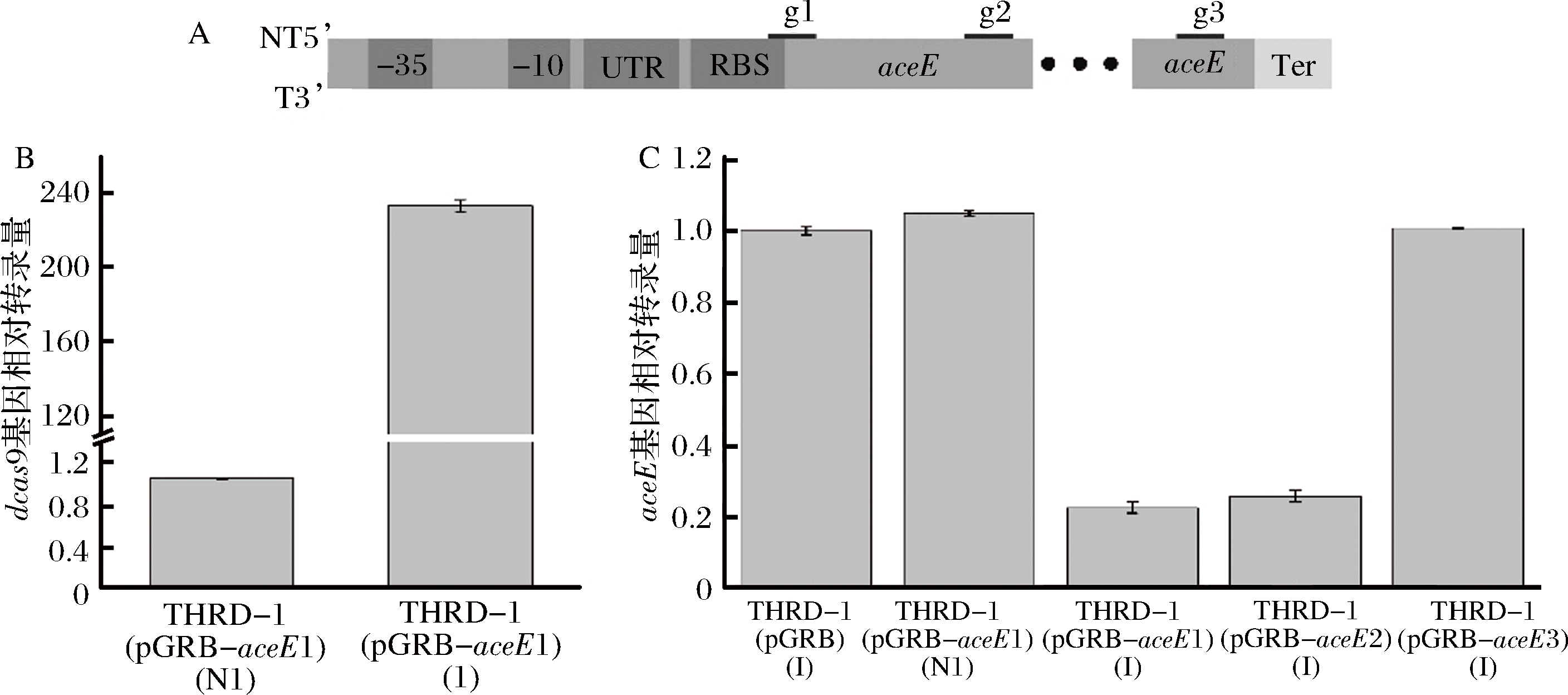
为了测试代谢干扰体系的可行性和阿拉伯糖诱导的严紧性,本实验构建了干扰aceE基因的pGRB质粒,并导入菌株THRD-1,检测阿拉伯糖诱导条件下的aceE基因和dcas9基因转录水平变化。前期研究报道[27],sgRNA设计与非模板链DNA链结合有较高的靶向效率,与模板链结合的sgRNA仅表现出较弱的靶向效率,且sgRNA靶向基因的ATG区域效果优于启动子区域,sgRNA靶向启动子区域与编码区中部效率相当。为了研究不同区域的sgRNA对基因转录的抑制作用,针对aceE基因构建了3个不同的靶向sgRNA序列,分别为aceE基因非模板链的ATG区域、编码区中部区域和编码区尾部区域(图1-A)。
通过RT-PCR对阿拉伯糖诱导条件下的基因dcas9进行转录分析,未诱导条件下dcas9基因的转录量定为1,经阿拉伯糖诱导后的dcas9表达量是未诱导条件下的233倍(图1-B),说明阿拉伯糖可以有效地诱导dcas9的表达。随后对阿拉伯糖诱导条件下的基因aceE进行转录分析,结果如图1-C所示。
对照菌株THRD-1(pGRB)中基因aceE的转录量定为1,菌株THRD-1(pGRB-aceE1) 在未诱导条件下基因aceE转录量没有降低,说明了阿拉伯糖诱导系统的严紧性。在阿拉伯糖诱导条件下,干扰ATG区域的菌株THRD-1(pGRB-aceE1)和干扰编码区中部区域的菌株THRD-1(pGRB-aceE2)中,基因aceE的转录量分别降低了77.78%和74.56%,干扰编码区尾部区对基因aceE转录没有效果。由此可以看出,抑制基因aceE转录强弱的顺序依次为:ATG区域、编码区中部区域和编码区尾部区域,与文献报道一致[28]。
2.3 干扰中心代谢基因对苏氨酸生产的影响
由图1的结果可以看出,利用基于阿拉伯糖诱导的CRISPRi干扰体系可以有效地调节目的基因的转录水平。为了提高苏氨酸的合成效率,本实验选择了中心代谢的关键基因zwf、pfkA、pykF、pck、aceE、gltA、sucA、sucC和sucD为干扰对象,分析9个基因的转录水平变化对苏氨酸合成的影响。每个靶向基因选择2个不同的干扰区域以实现不同的抑制水平,干扰的位置分别为对应基因的ATG区域和编码区中部区域。构建相应的干扰质粒并导入THRD-1,摇瓶发酵24 h,测定菌体生物量、葡萄糖消耗和苏氨酸产率,结果如表2所示。
表2 干扰中心代谢不同基因的菌株苏氨酸发酵结果
Table 2 Results of threonine fermentation with strains by interfering different central metabolic genes
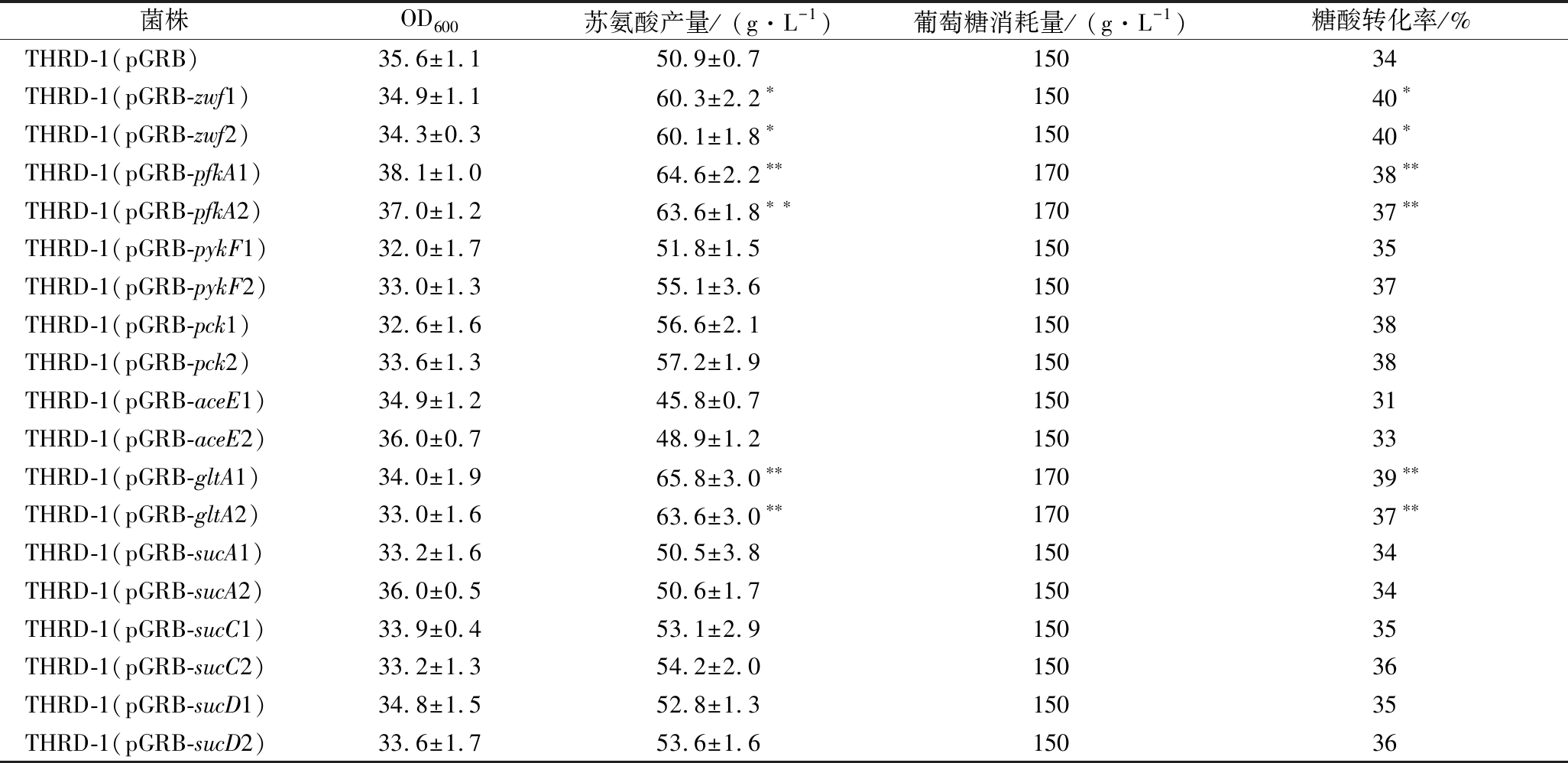
菌株OD600苏氨酸产量/ (g·L-1)葡萄糖消耗量/ (g·L-1)糖酸转化率/%THRD-1(pGRB)35.6±1.150.9±0.715034THRD-1(pGRB-zwf1)34.9±1.160.3±2.2∗15040∗ THRD-1(pGRB-zwf2)34.3±0.360.1±1.8∗15040∗ THRD-1(pGRB-pfkA1)38.1±1.064.6±2.2∗∗17038∗∗THRD-1(pGRB-pfkA2)37.0±1.263.6±1.8∗∗17037∗∗THRD-1(pGRB-pykF1)32.0±1.751.8±1.515035THRD-1(pGRB-pykF2)33.0±1.355.1±3.615037THRD-1(pGRB-pck1)32.6±1.656.6±2.115038THRD-1(pGRB-pck2)33.6±1.357.2±1.915038 THRD-1(pGRB-aceE1)34.9±1.245.8±0.715031 THRD-1(pGRB-aceE2)36.0±0.748.9±1.215033 THRD-1(pGRB-gltA1)34.0±1.965.8±3.0∗∗17039∗∗THRD-1(pGRB-gltA2)33.0±1.663.6±3.0∗∗17037∗∗THRD-1(pGRB-sucA1)33.2±1.650.5±3.815034THRD-1(pGRB-sucA2)36.0±0.550.6±1.715034THRD-1(pGRB-sucC1)33.9±0.453.1±2.915035THRD-1(pGRB-sucC2)33.2±1.354.2±2.015036 THRD-1(pGRB-sucD1)34.8±1.552.8±1.315035 THRD-1(pGRB-sucD2)33.6±1.753.6±1.615036
注:*表示0.01<P<0.05显著差异;**表示P<0.01差异极显著。
发酵结果显示,干扰pykF、pck、aceE、sucA、sucC和sucD基因的转录对菌株产酸和糖酸转化率方面没有明显的提升,说明干扰上述基因并不能提高苏氨酸生产菌的产酸效率。干扰zwf、pfkA和gltA基因对苏氨酸的产率和糖酸转化率都有不同程度的提高,这也证明可以采用CRISPRi技术来筛选对苏氨酸合成具有正向效果的基因。
zwf基因是葡萄糖6-磷酸进入HMP途径的关键基因,而HMP途径是苏氨酸合成的支路途径和生成CO2的主要途径。调节zwf基因的表达水平可以减缓HMP途径的代谢速率,减少碳源流失,使更多的碳源流向苏氨酸。干扰zwf基因的ATG区域和编码区中部区域,对应菌株苏氨酸产量分别为60.3和60.1 g/L,与对照菌株(50.9 g/L)相比,分别提高了18.5%和18.1%。发酵培养24 h,两者消耗葡萄糖的量均为150 g/L,糖酸转化率均为40%。
pfkA基因是EMP途径的关键基因,该基因编码的6-磷酸葡萄糖激酶是EMP途径的限速酶,调节pfkA 基因的转录水平,使EMP途径代谢速率变缓,减缓中心代谢速率,优化代谢网络,提高苏氨酸合成效率。干扰pfkA基因的ATG区域和编码区中部区域,对应菌株苏氨酸产量分别为64.6和63.6 g/L,与对照菌株(50.9 g/L)相比,分别提高了26.9%和25.0%。发酵培养24 h,消耗葡萄糖的质量浓度为170 g/L,糖酸转化率分别为38%和37%,与对照菌株34%相比分别提高了11.8%和8.8%。
gltA基因是乙酰辅酶A进入TCA循环的关键基因,调节gltA基因的转录水平,减缓TCA循环的代谢速率,进入TCA循环的草酰乙酸相对减少,更多的草酰乙酸进入苏氨酸合成途径,使苏氨酸的产率和糖酸转化率有一定程度的提高。干扰gltA基因的ATG区域和编码区中部区域,对应菌株的苏氨酸产量分别为65.8和63.6 g/L,与对照菌株(50.9 g/L)相比,分别提高了29.3%和25.0%。发酵培养24 h,消耗葡萄糖质量浓度为170 g/L,糖酸转化率分别为39%和37%,与对照菌株(34%)相比,分别提高了14.7%和8.8%。
依据上述结果分析,调节zwf、pfkA和gltA基因的表达水平,对应菌株的苏氨酸产率和糖酸转化率都有一定程度的提高。对同一基因而言,干扰基因ATG区域对应的菌株在苏氨酸产率和糖酸转化率方面均优于干扰基因编码区中部区域的菌株。对不同基因而言,调节pfkA和gltA基因的转录水平,均提高了苏氨酸的产率和糖酸转化率,且两者提升的幅度相同。虽然两者减缓了中心代谢速率,重新平衡了代谢网络,但重新平衡代谢网络后的菌株对葡萄糖的需求量有所增加,导致两者在糖酸转化率方面的提升幅度不及产酸方面。相比pfkA和gltA基因,调节zwf基因的转录水平,虽然在苏氨酸产率方面不及前两者,但由于在发酵培养过程中消耗葡萄糖的量相对较少,所以在苏氨酸糖酸转化率方面优于前者。同时,表2中菌株的OD600与对照菌株差别不大,表明调节基因的表达水平对菌体的生长没有显著影响。
3 结论
随着基因工程技术的不断发展,调节合成代谢途径基因的表达水平,优化合成代谢网络变得越来越重要。传统代谢工程研究中,为了增加目标产品的积累,通常通过删除相关基因,阻断支路途径来减少中间产物的消耗。然而,如果缺失的基因是必需的,细胞就会死亡或严重影响细胞生长。CRISPRi技术在转录层面调节目标基因的表达,避免了传统基因敲除方法的缺点,可以在不影响细胞生长的情况下调整和优化产物的合成代谢网络。
为了提高苏氨酸合成效率,本研究在苏氨酸工程菌建立了基于阿拉伯糖诱导CRISPRi干扰体系,并系统分析了中心代谢中关键基因表达水平的调节对苏氨酸合成的影响。结果表明,在阿拉伯糖的诱导条件下,通过调节zwf、pfkA和gltA基因转录水平,可以显著提高苏氨酸的产率和糖酸转化率。本研究为构建更高效的苏氨酸工程菌奠定了理论基础,也为其他生物制品的工程菌构建提供了实践参考。进一步的研究中,可以通过CRISPRi同时干扰多个靶基因,对苏氨酸合成代谢网络进行更好的调控,进一步提高苏氨酸产率和糖酸转化率。另外,还可以将对苏氨酸合成有益的相应sgRNA模块整合在基因组上,构建无质粒的工程菌。
参考文献
[1] LEE M H, LEE H W, PARK J H, et al. ImprovedL-threonine production ofEscherichia coli mutant by optimization of culture conditions[J]. Journal of Bioscience and Bioengineering, 2006, 101(2):127-130.
[2] 黄金, 徐庆阳,温廷益,等. 不同溶氧条件下L-苏氨酸生物合成菌株的代谢流量分析[J]. 微生物学报, 2008, 48(8):1 056-1 060.
[3] LEE K H, PARK J H, KIM T Y, et al. Systems metabolic engineering ofEscherichia coli forL-threonine production[J]. Molecular Systems Biology, 2007, 3(1):149-157.
[4] NOH M, YOO S M, KIM W J, et al. Gene expression knockdown by modulating synthetic small RNA expression inEscherichia coli[J]. Cell Systems, 2017, 5(40):418-426.
[5] YANG Y, LIN Y, LI L, et al. Regulating malonyl-CoA metabolism via synthetic antisense RNAs for enhanced biosynthesis of natural products[J]. Metabolic Engineering, 2015, 29:217-226.
[6] WANG H H, ISAACS F J, CARR P A, et al. Programming cells by multiplex genome engineering and accelerated evolution[J]. Nature, 2009, 460(7 257):894-898.
[7] MARRAFFINI L A, SONTHEIMER E J. CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea[J]. Nature Reviews Genetics, 2010, 11(3):181-190.
[8] YOO S M, NA D, LEE S Y. Design and use of synthetic regulatory small RNAs to control gene expression inEscherichia coli[J]. Nature Protocols, 2013, 8(9):1 694-1 707.
[9] NA D, YOO S M, CHUNG H, et al. Metabolic engineering ofEscherichia coli using synthetic small regulatory RNAs[J]. Nature Biotechnology, 2013, 31(2):170-174.
[10] GASIUNAS G, BARRANGOU R, HORVATH P, et al. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria[J]. Proceedings of the National Academy of Sciences of the United States of America, 2012, 109(39):2 579-2 586.
[11] SHEMIAKINA I I, ERMAKOVA G V, CRANFILL P J, et al. A monomeric red fluorescent protein with low cytotoxicity[J]. Nature Communications, 2012, 3:1 204.
[12] GARNEAU J E, DUPUIS M E, VILLION M, et al. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA[J]. Nature, 2010, 468(7 320):67-71.
[13] MOJICA F J, DIEZVILLASENOR C, GARCIA-MARTINEZ J, et al. Short motif sequences determine the targets of the prokaryotic CRISPR defence system[J]. Microbiology, 2009, 155(3):733-740.
[14] JINEK M, CHYLINSKI K, FONFARA I, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity[J]. Science, 2012, 337(6 096):816-821.
[15] JIANG W, BIKARD D, COX D, et al. RNA-guided editing of bacterial genomes using CRISPR-Cas systems[J]. Nature Biotechnology, 2013, 31(3):233-239.
[16] QI L, LARSON M H, GILBERT L A, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression[J]. Cell, 2013, 152(5):1 173-1 183.
[17] DOMINGUEZ A A, LIM W A, QI L S. Beyond editing: Repurposing CRISPR-Cas9 for precision genome regulation and interrogation[J]. Nature Reviews Molecular Cell Biology, 2016, 17(1):5-15
[18] WU J, DU G, CHEN J, et al. Enhancing flavonoid production by systematically tuning the central metabolic pathways based on a CRISPR interference system inEscherichia coli[J]. Scientific Reports, 2015, 5:13 477
[19] CLETO S, JENSEN J K, WENDISCH V F, et al.Corynebacterium glutamicum metabolic engineering with CRISPR interference (CRISPRi)[J]. ACS Synthetic Biology, 2016, 5(5):375-385.
[20] KIM S K, HAN G H, SEONG W, et al. CRISPR interference-guided balancing of a biosynthetic mevalonate pathway increases terpenoid production[J]. Metabolic Engineering, 2016, 38:228-240
[21] YAO L, CENGIC I, ANFELT J, et al. Multiple gene repression inCyanobacteria using CRISPRi[J]. ACS Synthetic Biology, 2016, 5(3):207-212.
[22] LI Y, LIN Z, HUANG C, et al. Metabolic engineering ofEscherichia coli using CRISPR-Cas9 meditated genome editing[J]. Metabolic Engineering, 2015, 31:13-21.
[23] BAO Z, XIAO H, LIANG J, et al. Homology-integrated CRISPR-Cas (HI-CRISPR) system for one-step multigene disruption inSaccharomyces cerevisiae[J]. ACS Synthetic Biology, 2015, 4(5):585-594.
[24] COBB R E, WANG Y, ZHAN H. High-efficiency multiplex genome editing ofStreptomyces species using an engineered CRISPR/Cas system[J]. ACS Synthetic Biology, 2015, 4(6):723-728.
[25] GUZMAN L M, BELIN D, CARSON M J, et al. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter[J]. Journal of Bacteriology, 1995, 177(14):4 121-4 130.
[26] NAKASHIMA N, TAMURA T. A new carbon catabolite repression mutation ofEscherichia coli,mlc*, and its use for producing isobutanol[J]. Journal of Bioscience and Bioengineering, 2012, 114(1):38-44.
[27] FU Y, FODEN J A, KHAYTER C, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells[J]. Nature Biotechnology, 2013, 31(9): 822-826.
[28] LYU L, REN Y L, CHEN J C, et al. Application of CRISPRi for prokaryotic metabolic engineering involving multiple genes, a case study: Controllable P(3HB-co-4HB) biosynthesis[J]. Metabolic Engineering, 2015, 29:160-168.
Threonine synthesis under interfered transcriptions of genes involved in central metabolic pathway by CRISPRi
LIU Xufeng1, WANG Ning1, HAO Yanan1, LI Yingzi1, FAN Xiaoguang1, 2, XIE Xixian1, 2*
1(National and Local United Engineering Lab of Metabolic Control Fermentation Technology (Tianjin University of Science and Technology), Tianjin 300457, China) 2(Tianjin Engineering Research Center of Microbial Metabolism and Fermentation Process Control, Tianjin 300457, China)
Abstract Threonine is an important feed amino acid with increasing demand. Improving threonine productivity and glucose conversion rate and reducing the production costs have become an important subject. In this study,Escherichia coli THRD, a threonine-producing strain, was used to study the effects of different transcription levels of nine genes involved in central metabolic pathway on threonine synthesis by using clustered regularly interspaced short palindromic repeats interference (CRISPRi). The results showed that the transcriptional interference of geneszwf,pfkA, andgltA improved the synthesis efficiency of threonine. The threonine titers of corresponding strains were 60.3, 64.6, and 65.8 g/L, respectively, which were increased by 18.5%, 26.9%, and 29.3%, respectively, compared with the original strain (50.9 g/L). The glucose conversion rates were 40%, 38%, and 39%, respectively, which were 17.7%, 11.8%, and 14.7% higher than that of the original strain (34%). The results showed that by modifying transcriptional levels of genes that involved in central metabolism by CRISPRi could regulate the cellular metabolic network and redirect more carbon flux to threonine, resulted improved synthesis efficiency of threonine. Overall, this study provides a reference for constructing other bioengineered bacteria.
Key words Escherichia coli; threonine; metabolic interference; glucose conversion rate; clustered regularly interspaced short palindromic repeats interference (CRISPRi)
第一作者:硕士研究生(谢希贤教授为通讯作者,E-mail:xixianxie@tust.edu.cn)。
基金项目:国家自然科学基金青年基金(31700037);工业微生物优良菌种选育与发酵技术公共服务平台项目(17PTGCCX00190)
收稿日期:2018-11-29,改回日期:2019-01-17
DOI:10.13995/j.cnki.11-1802/ts.019489