启动子串联及改造提高FAD为辅基的葡萄糖脱氢酶在Bacillus subtilis中的表达
张玲1,林荣2,宋祖坤1,王男1,杨海麟1*
1(江南大学,工业生物技术教育部重点实验室,江苏 无锡, 214122) 2(苏州盛迪亚生物医药有限公司,江苏 苏州, 215000)
摘 要以黄素腺嘌呤二核苷酸(flavin adenine dinucleotide,FAD)为辅基的葡萄糖脱氢酶(glucose dehydrogenase with FAD,FAD-GDH,EC 1.1.99.10),与辅基结合紧密,催化效率高,是临床检测血糖指标的新型诊断用酶。将Burkholderia cepacia的FAD-GDH基因(gdh)构建含单启动PHpaⅡ的穿梭质粒pMA5-1,在蛋白酶缺陷型菌株Bacillus subtilis WB600中表达。为了获得该酶的高效表达,采用启动子串联及改造策略考察产酶情况。将4种启动子(PamyQ’,P43,PgsiB,Popuaa)分别与质粒上自带的启动子PHpaⅡ串联,结果表明PHpaⅡ-PamyQ’串联组合获得的FAD-GDH胞内酶活最高,为2 497 U/L,是串联前单启动子的2.7倍。为了减少发酵过程中,葡萄糖和甘油对产酶的抑制作用,在串联组合的基础上删去PamyQ’启动子中与碳代谢调控蛋白结合的cre位点,使胞内产酶水平提高至3 626 U/L,说明cre位点的去除能够减少碳代谢产物对启动子转录的抑制。本研究为新型诊断用酶FAD-GDH的菌种改造和工业化生产应用提供参考与借鉴。
关键词黄素腺嘌呤二核苷酸(FAD);葡萄糖脱氢酶;枯草芽孢杆菌;串联启动子;cre位点
葡萄糖氧化酶(glucose oxidase,GOD)被广泛应用于血糖指标的临床生化检测。近些年发现葡萄糖脱氢酶(glucose dehydrogenase,GDH)具有替代葡萄糖氧化酶的应用潜力,因其不以氧气为电子受体,不受溶解氧的限制,检测结果误差更小[1-2]。葡萄糖脱氢酶按照辅基或辅酶差异可分为[3]:(1)以烟酰胺腺嘌呤二核苷酸(hicotinamide adenine dinucleotide, NAD(P)+)为辅酶的葡萄糖脱氢酶(NAD(P)-GDH,EC 1.1.1.47)。(2)以吡咯并喹啉醌(pyvrolidine quinoline quinone, PQQ)为辅基的葡萄糖脱氢酶(PQQ-GDH,EC 1.1.5.2)。(3)以黄素腺嘌呤二核苷酸(flavin adenine dinucleotide,FAD)为辅基的葡萄糖脱氢酶(FAD-GDH,EC 1.1.99.10)。其中FAD-GDH以辅基结合紧密,催化效率较高,可作为替代目前诊断用葡萄糖氧化酶的首选。
具有活性的FAD-GDH可能为单体[4]或同源二聚体[5]。FAD-GDH主要来源于丝状真菌[6],如:Penicillium sp.[7],Mucor sp.[8],Aspergillus sp.[9-10],Glomerella cingulata[11]等,重组表达和纯化较为困难。来源于Glomerella cingulata的FAD-GDH在E.coli中重组表达,常常需要与分子伴侣系统共表达才能实现可溶性表达,蛋白纯化和放大生产较为困难[11]。在酵母菌中表达量较高,但存在糖基化修饰,会干扰血糖检测电极的灵敏度,实际生产应用中需进行酶的去糖基化步骤[12]。
FAD-GDH罕见于细菌。本团队先前尝试将来源于细菌Burkholderia cepacia的FAD-GDH基因(gdh)在大肠杆菌中表达,该酶热稳定性好,Tm值可达77 ℃。为了探索该基因在其他宿主的表达情况,本文尝试将来源于原核Burkholderia cepacia的gdh基因在枯草芽孢杆菌表达系统中表达。枯草芽孢杆菌作为表达宿主,遗传背景清楚,对培养基要求较低,生长快速,不产内毒素,适用于原核生物来源重组蛋白的分泌表达。
筛选高效的启动子可实现外源基因在枯草芽孢杆菌中高效表达。启动子依据是否需要诱导分为:(1)组成型启动子,如P43,PHpaⅡ,PamyQ’;(2)诱导型启动子,如Popuaa,Pgsib。组成型的启动子能在菌体内持续进行基因表达,受环境因素的影响较少,或变化很小。诱导型启动子在诱导因子存在下,相应的基因被激活,基因表达,积累目的产物[13]。许多研究表明双启动子相比单启动子更能促进外源基因的表达。重组菌株B.subtilis CCTCCM 2016536用于表达β-环糊精糖基转移酶,双启动子PHpaⅡ-PamyQ’表达系统的表达量是单个启动子PamyQ’表达系统表达量的1.3倍,主要由于双启动子下外源基因转录水平高于单启动子[14]。启动子PAmyR2和启动子Pblma分别插入PHpaII启动子的下游,4-α-葡聚糖转移酶的表达量和单启动子PHpaII比较分别提高11、12倍[15]。双启动子PHpaII-PgsiB表达氨肽酶分别是单个PHpaII和PgsiB启动子表达的2.3和2.2倍[16]。
将启动子突变改造也是提高枯草芽孢杆菌产物积累的有效办法。碳源代谢在菌体发酵过程中占有重要地位,表达外源产物的过程中碳代谢产物例如葡萄糖等,会对目的产物的生成产生抑制作用,即碳代谢产物抑制(carbon catabolite repression,CCR),这种抑制作用主要是通过碳代谢控制蛋白A (carbon catabolite protein A,CcpA) 复合物结合到启动子区域的cre位点抑制转录[17]。cre位点的突变可有效地减少CcpA介导的碳代谢抑制作用。NICHOLSON等通过随机突变筛选得到抗碳代谢产物的淀粉酶生产菌株,经过测序发现amyE启动子的cre序列中GC碱基突变成AT[18]。NAGARAJAN等克隆来自高产淀粉酶的Bacillus subtilis KCC103菌株的启动子amyR4,该启动子能够有效地抵抗碳代谢产物的抑制,实现异源蛋白的高效表达,序列研究表明启动子转录起始位点上游cre序列中+4(C-T),-7(T-A)碱基发生突变[19]。
本研究尝试将Burkholderia cepacia的FAD-GDH基因(gdh)在蛋白酶缺陷型菌株B.subtilis WB600中进行表达,通过启动子筛选、串联及改造等策略,以期获得FAD-GDH高表达量。
1 材料与方法
1.1 材料与试剂
1.1.1 菌株
葡萄糖脱氢酶基因(gdh)由南京金斯瑞合成,构建于载体pUC57;E.coli JM109,B.subtilis 168,B.subtilis WB600菌株由实验室保存(表1)。
1.1.2 主要试剂
限制性内切酶、T4 DNA连接酶、rTaq DNA聚合酶、蛋白质Low Marker、DNA Marker均购自大连宝生物有限公司;PCR产物回收试剂盒;DNA凝胶回收试剂盒购自上海生工股份有限公司;其他所用试剂均购于国药集团药业股份有限公司。
表1 实验所用构建菌株及质粒
Table 1 Strains and plasmids constructed in this study

质粒及菌株特征来源pUC57-gdhpUC57派生,gdh基因南京金斯瑞pMA5B. subtilis-E. coli穿梭载体,KnR,ApR实验室保存pMA5-1pMA5派生,gdh基因本实验构建pMA5-11pMA5-1派生PHPaⅡ-PamyQ’本实验构建pMA5-12pMA5-1派生PHPaⅡ-P43本实验构建pMA5-13pMA5-1派生PHPaⅡ-PgsiB本实验构建pMA5-14pMA5-1派生PHPaⅡ-Popuaa本实验构建pMA5-2pMA5-2派生PHPaⅡ-PamyQ2本实验构建WB1转化质粒pMA5-1后获得本实验构建WB11转化质粒pMA5-11后获得本实验构建WB12转化质粒pMA5-12后获得本实验构建WB13转化质粒pMA5-13后获得本实验构建WB14转化质粒pMA5-14后获得本实验构建WB2转化质粒pMA5-2后获得本实验构建
1.1.3 培养基及缓冲液
(1) LB培养基(g/L):酵母粉5.0,蛋白胨10.0,NaCl 5.0,加入2%(质量分数)的琼脂即为LB固体培养基。(2) TB培养基(g/L):甘油4.0,酵母粉24.0,蛋白胨12.0,溶解在800 mL去离子水中,高压灭菌后冷却至40 ℃加入200 mL灭菌的缓冲液(K2HPO4 16.4 g,KH2PO4 2.32 g,溶解于去离子水中,定容至200 mL,0.22 μm的滤膜过滤除菌或高压灭菌)。(3) 抗 生素溶液:先配置成质量浓度为100 mg/mL的母液,然后用0.22 μm的滤膜过滤除菌,用小的EP管分装,置于-20 ℃ 保存。(4) 磷酸钾缓冲液(phospate buffered,PB,50 mmol/L, pH 7.0):取86 mL的0.1 mol/L K2HPO4,加入14 mL的0.1 mol/L KH2PO4,再加入100 mL水,调节pH至7.0。
1.2 方法
1.2.1 重组质粒pMA5-1的构建
以质粒pUC57-gdh为模板进行PCR扩增,引入BamHⅠ/MluⅠ位点,扩增产物回收双酶切,与同样双酶切的质粒pMA5连接,构建表达质粒pMA5-1。
1.2.2 串联启动子重组质粒的构建
以B.subtilis 168的DNA为模板,利用引物PamyQ’-F/PamyQ’-R,P43-F/P43-R,PgsiB-F/PgsiB-R, Popuaa-F/ Popuaa-R,分别PCR扩增相应的启动子序列,同时引入NdeⅠ/MluⅠ位点,双酶切启动子序列与同样双酶切的质粒pMA5-1连接,构建双启动子质粒pMA5-1n(表2)。
1.2.3 培养方法
种子液的制备:从卡纳抗性的固体培养基上,挑取重组菌B.subtilis单菌落,接种到LB液体培养基,37 ℃,200 r/min摇床培养8 h。
表2 实验所用引物
Table 2 Oligonucleotides used in this study

引物序列(5’-3’)GDH-FAATTCCGGATCC ATGCACAACGACAACACCGDH-RAATAACGCGTGGTTAGTGGTGATGGTGATGPamyQ’ -FACTACATATGGGCGGCGTTCTGTTTCTGCTTCGGTATPamyQ’ -RAATTGGATCCATAAGGCAGTAAAGAGGTTTTGAP43-FATTACATATGTGATAGGTGGTATGTTTTCGCTTGAACP43-RACGGATCCGTGTACATTCCTCTCTTACCTATAAPgsiB-FACCTCATATGCTATCGAGACACGTTTGGCTGGAAAAAPgsiB-RCCAGGATCCTTCCTCCTTTAATTGGTGTTGGTTGTPopuaa-FATACATATGAGGTAGGAGAAAAATCACCCGTAAAAPopuaa-RATTGGATCCCATATTTTCCCTCCATATTAAGCAATCPamyQ’.del.14nt-FGTGATTGTGATAATTTTAAAAAATTCTCCAGTCTPamyQ’.del.14nt-RAGACTGGAGAATTTTTTAAAATTATCACAATCAC
摇瓶发酵培养:以体积分数4%转接种子液于发酵培养基中,30 ℃,220 r/min,培养48 h。
1.2.4 菌体生物量测定
取20 mL不同浓度的菌液,8 000 r/min离心10 min, 用pH 7.0,50 mmol/L的PB缓冲液清洗2次,测定湿重,105 ℃烘干至恒定值,称取干重。菌体湿重约是干重的3.8倍。
1.2.5 葡萄糖脱氢酶酶活测定及SDS-PAGE分析
酶活定义:在一定条件下,每分钟还原1 μmol DCPIP所需的酶量定义为一个酶活单位。
取100 μL适当稀释的酶液,加入3 mL显色液(50 mmol/L pH 7.0的PB缓冲液, 0.03 mmol/L的DCPIP,1.6 mmol/L的PMS,100 mmol/L的葡萄糖溶液)轻轻混匀,用重蒸水调零,37 ℃每隔1 min记录600 nm处吸光值的变化,共记录2~3 min。
用酶的缓冲液代替酶液作为对照。
采用5%的浓缩胶与15%的分离胶,pH 8.3的Tris-Gly电泳缓冲液,90 V恒压,将剥离电泳后的胶,染色,脱色。胶的具体配置操作方法参照SDS-PAGE试剂盒。
1.2.6 基质辅助激光解析电离串联飞行时间质谱分析
样品处理:(1)用刀片切下胶上目标条带,放入离心管中;(2)加入400 μL脱色液脱色,清洗至透明,去除上清,冻干或加50 μL乙腈,放置5 min,去除乙腈,重复1次,晾干,胶粒应为白色;(3)加入90 μL 100 mmol/L NH4HCO3,10 μL 100 mmol/L DTT,56 ℃ 放置30 min; (4)去上清,加100 μL 100%乙腈,5 min后吸去;(5)加入70 μL 100 mmol/L NH4HCO3,30 μL 200 mmol/L IAA,暗处理20 min;(6)去上清,加入100 μL 100 mmol/L NH4HCO3,室温放置15 min;(7)去上清,加入100 μL 100%乙腈,5 min后吸去,冻干或加50 μL乙腈,放置5 min,去除乙腈,重复1次,晾干,胶粒应为白色;(8)加入6 μL酶液,4 ℃放置30~60 min,使胶块充分吸涨;(9)加入20 μL 25 mmol/L NH4HCO3缓冲液,37 ℃放置20 h左右;(10)取0.6 μL酶解液点样,自然晾干,覆盖0.6 μL基质,晾干。进行上机质谱分析,并将得到的结果导入NCBI网站比对。
2 结果与分析
2.1 重组质粒pMA5-1的构建表达及蛋白质谱鉴定
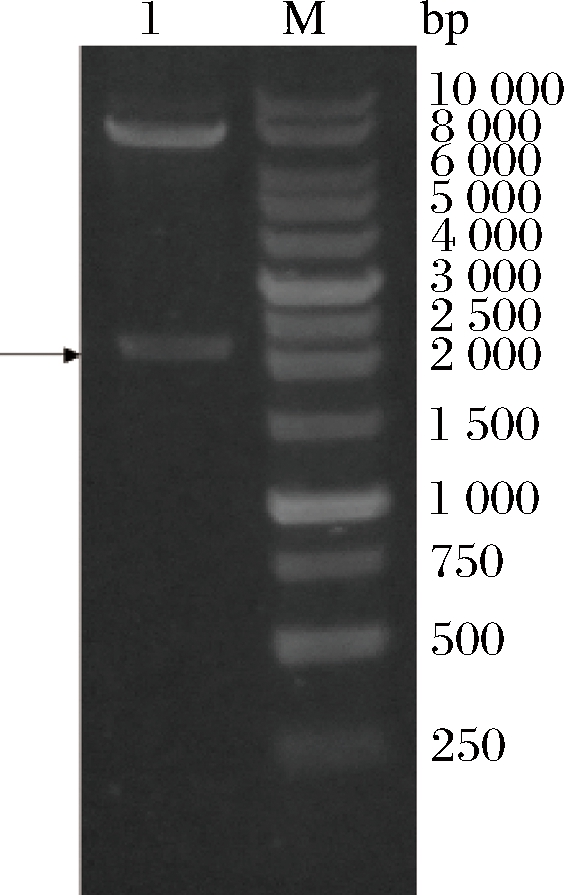
将gdh基因构建到pMA5上转入B.subtilis,研究是否能够表达具有活性的FAD-GDH。将pMA5-1转化E.coli JM109,经过氨苄抗性平板筛选,从阳性转化子中提取质粒,利用BamHⅠ和MluⅠ双酶切电泳验证,验证结果如图1,凝胶电泳条带与目的基因大小一致,并将验证正确的基因测序,与目的序列比对,结果表明构建成功。
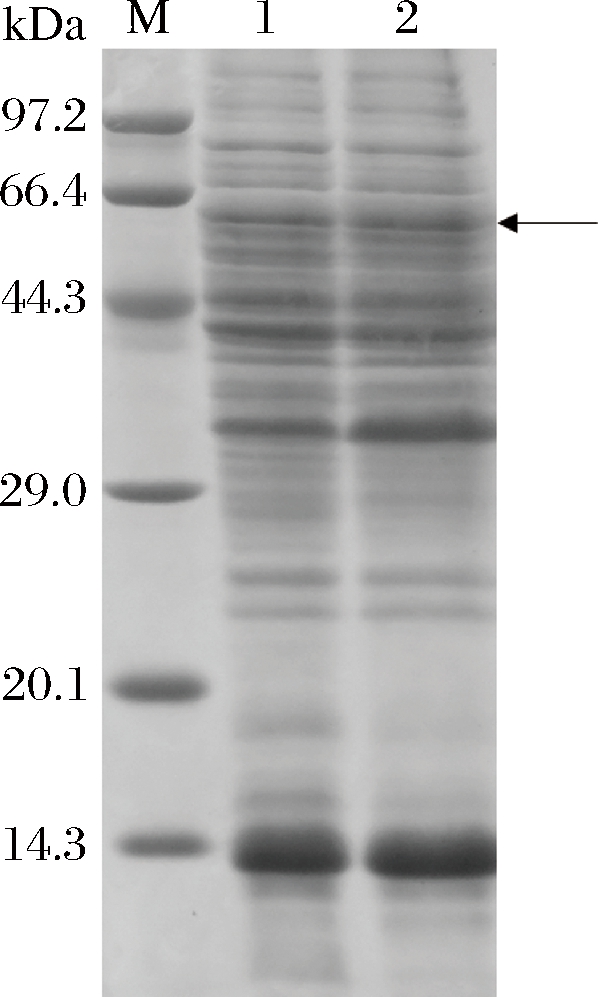
将构建成功的质粒pMA5-1转入B.subtilis感受态WB600,从含有卡纳抗性的平板上挑选阳性克隆,获得重组菌株WB1,将获得的重组菌摇瓶发酵培养48 h,菌体离心收集,细胞破碎,上清离心,测得酶活为962 U/L,并将离心获得的上清取30 μL,加入10 μL 4×SDS-PAGE的上样缓冲液,煮沸10 min,进行SDS-PAGE检测,结果见图2,由于空白对照处也存在一条60 kDa的条带,所以将WB1破壁上清SDS-PAGE图谱中60 kDa处的条带切胶回收,进行飞行质谱分析,比对结果显示与目的条带匹配度最高的为来源于Burkholderia cepacia的葡萄糖脱氢酶,分子质量60 kDa,进一步表明FAD-GDH在B.subtilis WB600中表达成功。
2.2 串联启动子提高FAD-GDH的表达
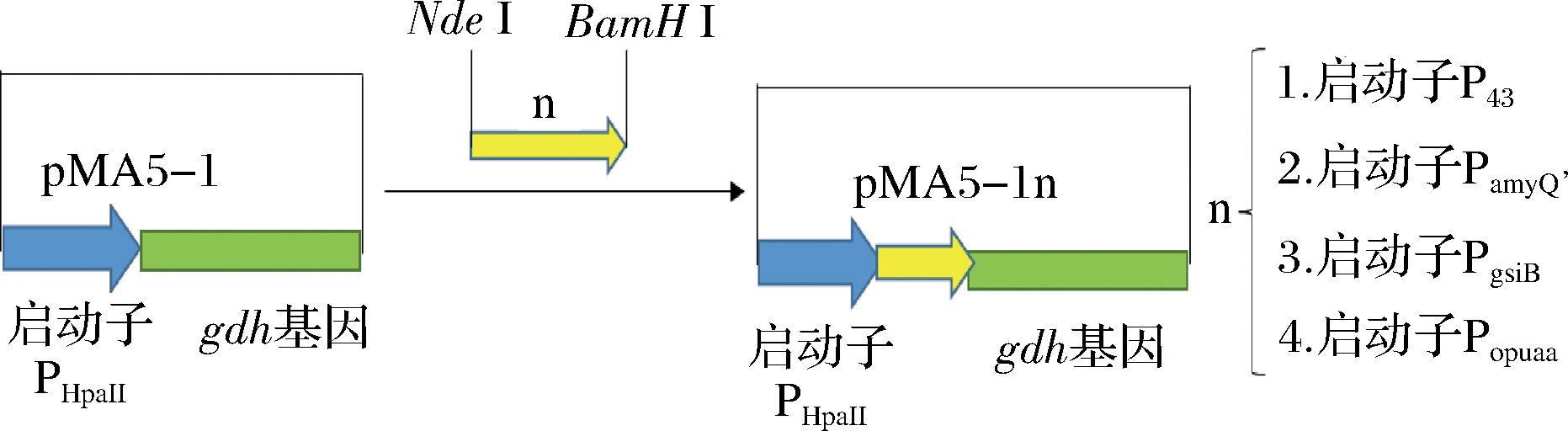
采用双启动子串联策略来提高FAD-GDH的表达量,本文选取的启动子见表3。启动子PHpaⅡ是质粒pMA5上自带的启动子,本文选取其他4种启动子串联在PHpaⅡ下游构建含有双启动子。其中PamyQ’和P43为组成型的启动子,具有强启动性、无需添加诱导剂可直接产生目的蛋白的优势;PgsiB和Popuaa为盐诱导型启动子,受盐浓度激活调控目的蛋白大量产生的特点。4种双启动子表达质粒pMA5-11(PHpaII-PamyQ’),pMA5-12(PHpaII-P43),pMA5-13(PHpaII-PgsiB),pMA5-14(PHpaII-Popuaa)构建流程见图3。
以B.subtilis 168基因组为模板进行PCR扩增相应的启动子序列,扩增产物回收双酶切插入启动子PHpaII的下游,分别获得重组质粒pMA5-11,pMA5-12,pMA5-13,pMA5-14。利用NdeⅠ和MluⅠ双酶切电泳验证,验证结果如图4,并将验证正确的基因测序,与目的序列比对,比对结果表明构建成功。
分别将构建成功的质粒pMA5-11,pMA5-12,pMA5-13,pMA5-14,转化感受态B.subtilis WB600,从含有卡纳抗性的平板上挑选阳性克隆,获得菌株WB11,WB12,WB13,WB14,以及含有单启动子PHpaⅡ的重组菌WB1,将获得的上述重组菌与含有单启动子PHpaⅡ的重组菌WB1分别摇瓶发酵培养48 h,菌体离心收集,细胞破碎。
表3 本文所构建的来源于B.subtilis的启动子
Table 3 The promoter fromB.subtilis used in this study

启动子来源特点参考文献PHpaⅡStaphlococcus aureus组成型启动子,受到RNA合成的刺激[20]PamyQ’B. subtilis受到DegS-DegU系统的调节[21]P43B. subtilis胞苷/脱氧胞苷脱氨酶启动子,介导含有σA或σB RNA聚合酶的转录[22-23]PgsiBB. subtilis受到σB因子调节,对温度,盐,乙醇,葡萄糖匮乏敏感[24-25]PopuaaB. subtilis盐诱导型启动子[26]
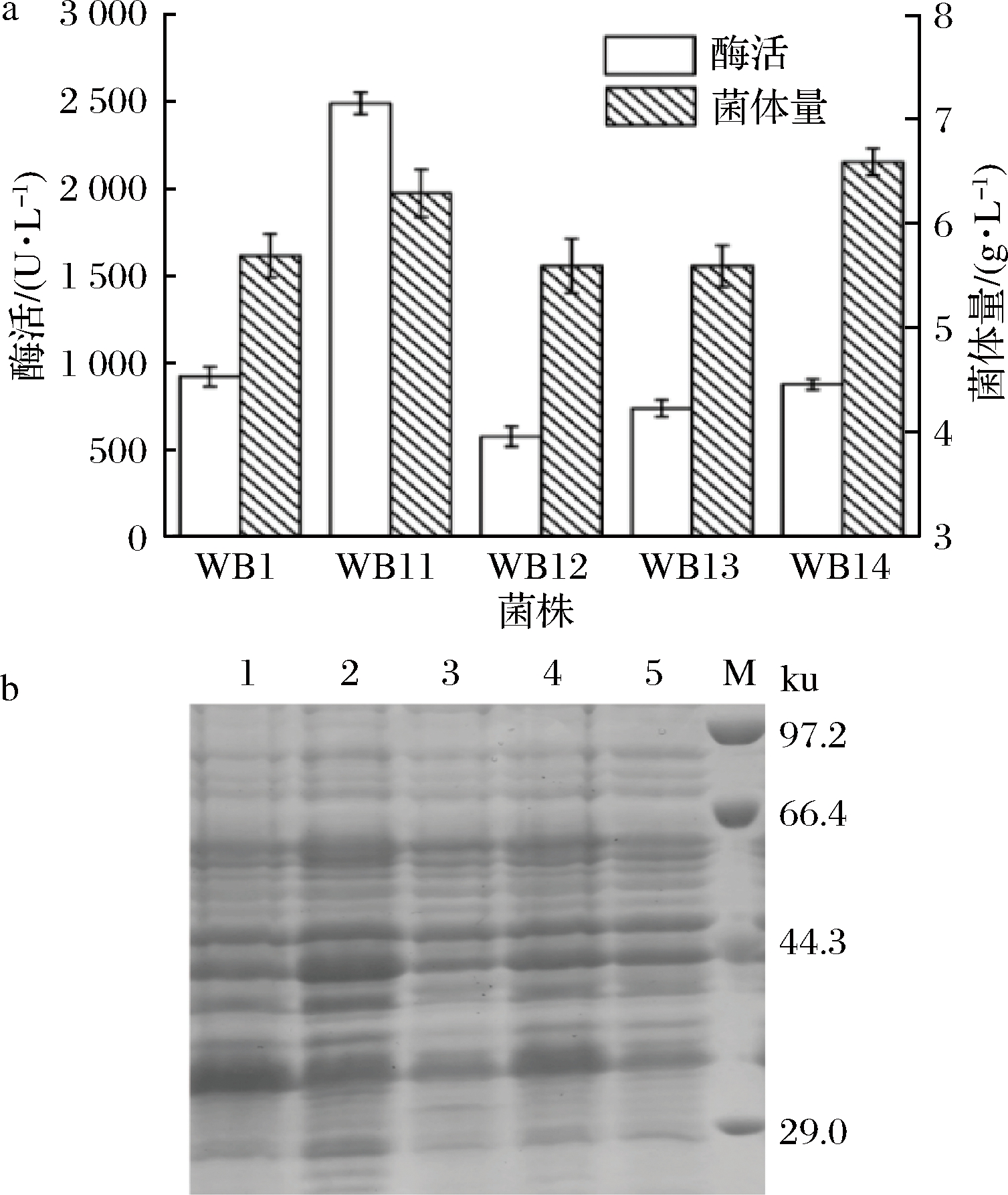
不同启动子的强度通过测定菌体胞内酶活来衡量。PamyQ’和P43为组成型的启动子[21-23],PgsiB和Popuaa为盐诱导型启动子[24-26],受盐浓度激活,可利用质量浓度为4 g/L的NaCl进行诱导表达。含单启动子PHpaⅡ重组菌株WB1及4种双启动子重组菌WB11,WB12,WB13,WB14的胞内酶活分别为924、2 497、 578、740、878 U/L(图5-a)。其中含有组成型启动子PamyQ’的双启动子重组菌WB11 (PHpaⅡ-PamyQ’)胞内酶活最高,约是酶活最低的重组菌株WB12的4倍,是单启动子PHpaⅡ表达FAD-GDH酶活的2.7倍。分别取30 μL上述离心获得的上清加入10 μL 4×的上样缓冲液,煮沸10 min,进行SDS-PAGE检测,结果见图5-b, 重组菌发酵液在60 kDa处都有目的蛋白产生。
2.3 删除启动子PamyQ’上cre位点促进FAD-GDH的发酵产酶
上述研究中表明最佳的串联双启动子PHpaII-PamyQ’使酶活提高到2 497 U/L,具有促进该酶表达的效果,但有研究表明PamyQ’启动子在发酵过程中会受到葡萄糖浓度的抑制,随着葡萄糖浓度的增加会明显抑制产酶[14]。本研究先考察葡萄糖是否会严重抑制串联启动子PamyQ’突变菌株的发酵产酶。比较不同浓度的葡萄糖和甘油条件下该突菌变株的发酵产酶情况。
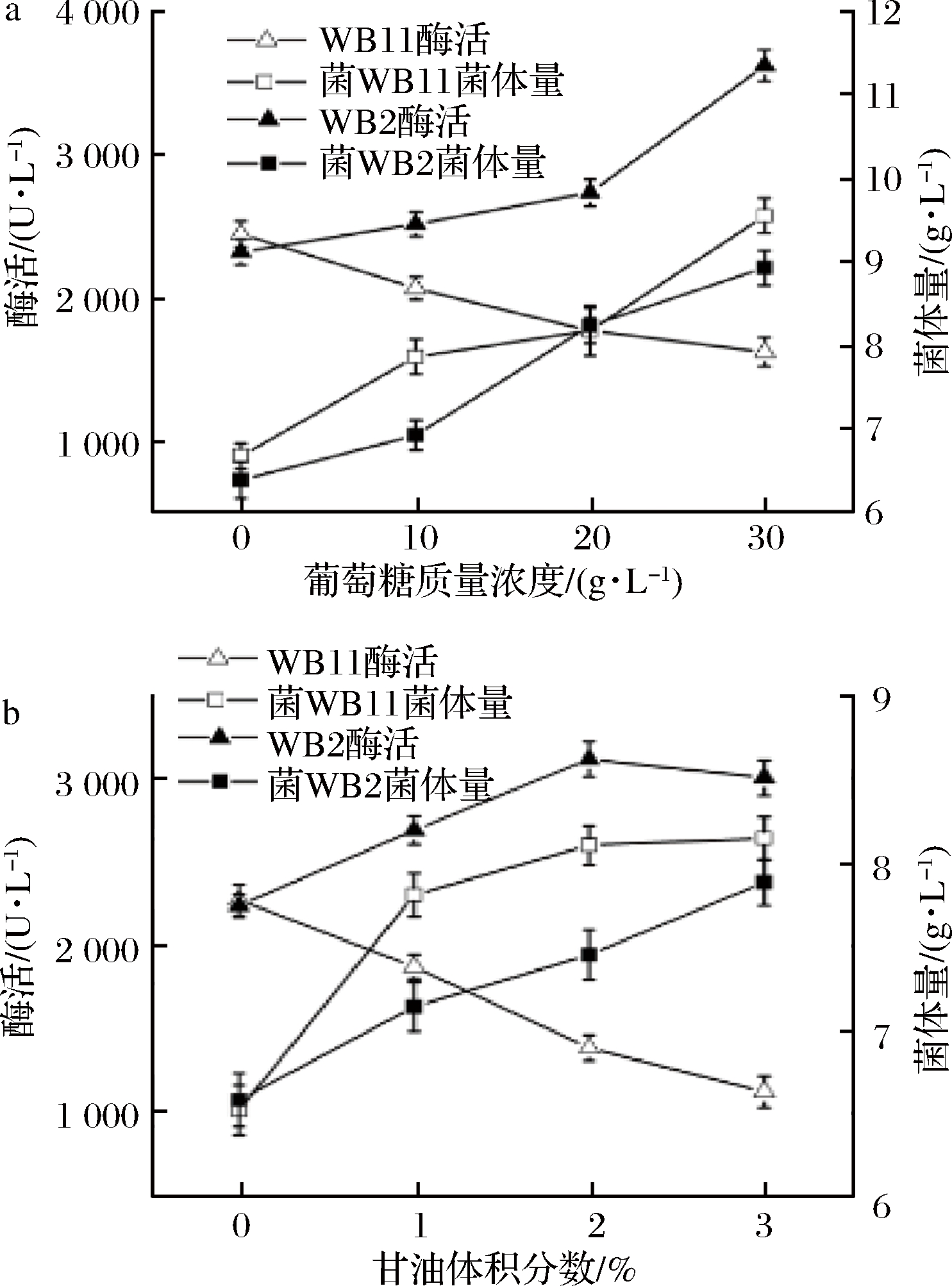
将改造前重组菌WB11(含有双启动子PHpaII-PamyQ’)发酵培养48 h,收集菌体,酶活验证是否存在碳代谢产物抑制效应。结果如图6,通过测定酶活检测FAD-GDH的表达情况,随着培养基碳源的葡萄糖或甘油浓度的增加,菌体量上升,酶活逐渐下降。未添加葡萄糖,菌体胞内酶活是添加30 g/L的葡萄糖培养菌体胞内酶活的1.5倍;当未添加甘油,胞内酶活是添加3%(体积分数)的甘油胞内酶活的2倍,说明串联启动子PamyQ’后碳源浓度增加会抑制产酶。
枯草芽孢杆菌中,碳代谢产物的抑制效应主要是通过碳代谢控制蛋白CcpA,结合到启动子区域的cre位点抑制转录。cre位点是一段含有回文结构,存在一些碱基保守的序列。CcpA如何与多样的cre位点结合,具体的机制还不清楚,但改造和移除该位点可有效减少碳代谢的抑制[18-19]。
为了减少碳代谢产物的抑制效应,基于双启动子PHpaII-PamyQ’,通过重叠延伸PCR[27]移除PamyQ’启动子中14个核苷酸的cre位点,构建双启动子PHpaII-PamyQ2,转入B.subtilis WB600,构建WB2重组菌,发酵培养基添加30 g/L的葡萄糖,菌体胞内酶活力达3 626 U/L, 约是启动子改造前添加30 g/L的葡萄糖菌体胞内酶活的2.2倍。如图7-b重组菌株WB2发酵培养基中添加2%(体积分数)的甘油菌体胞内酶活力达3 119 U/L,是改造之前重组菌株WB11添加2%的甘油菌体胞内酶活的2.1倍。重组菌株WB111发酵培养中添加3%的甘油胞内酶活比添加2%的甘油有所下降,可能是过多的碳源导致菌体生长过快,抑制重组蛋白表达。
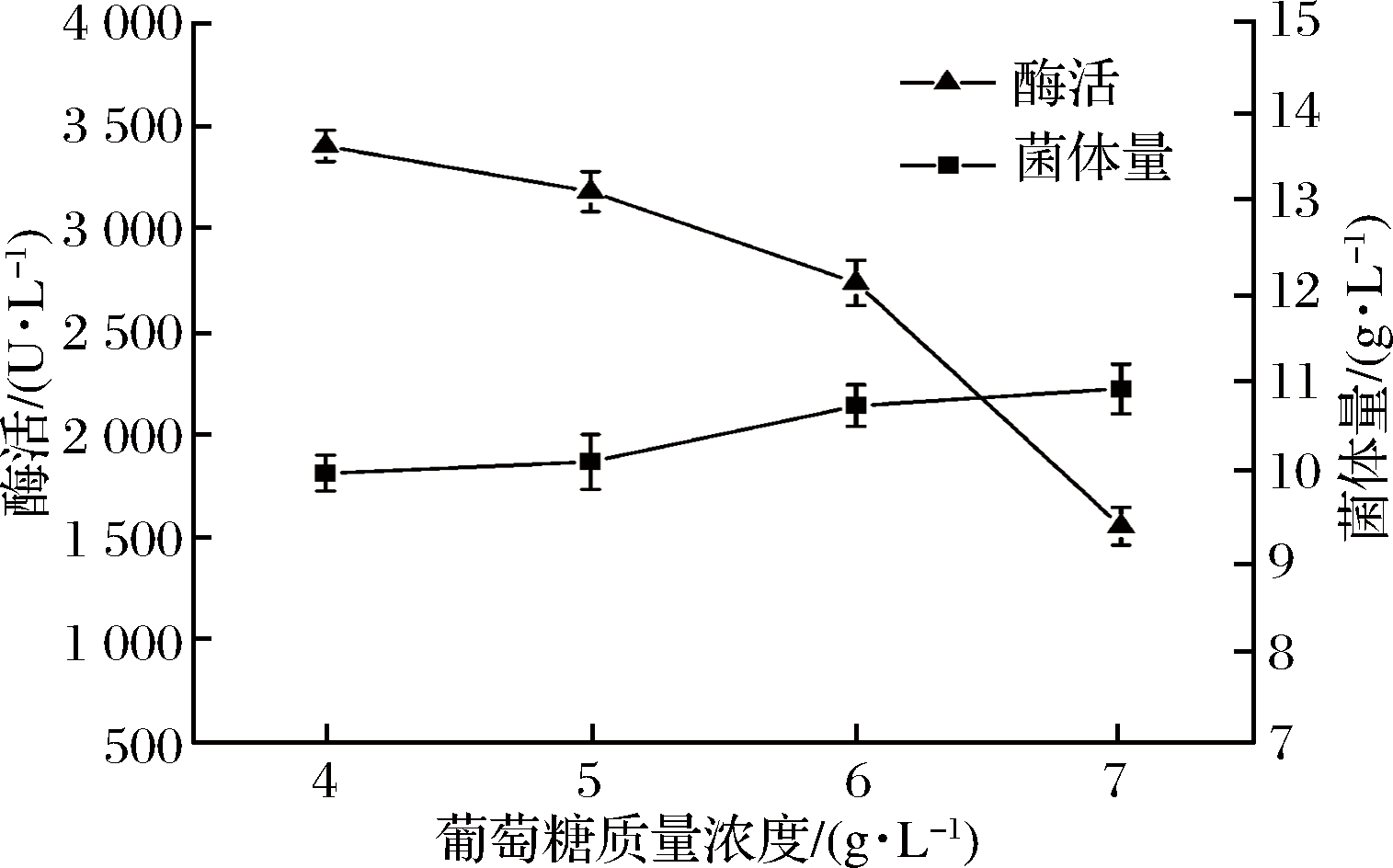
为了验证改造后的重组菌WB2,培养时添加高浓度的葡萄糖是否会抑制FAD-GDH的表达,向培养基中分别添加40、50、60、70 g/L的葡萄糖,测定破碎菌体上清中的酶活及菌体干重,结果如图7,随着葡萄糖浓度的上升,菌体干重趋于稳定,可能是菌体生长受到溶解氧浓度的限制,胞内酶活随着葡萄糖浓度上升而下降,过快的生长速度导致质粒丢失。培养基添加30~40 g/L的葡萄糖,一定程度上促进菌体生长及酶的表达,删去启动子PamyQ’的cre位点可减少碳源代谢产物对基因转录的抑制,促进外源基因表达。
3 结论
构建含有单启动子PHpaⅡ的重组质粒pMA5-1,转入B.subtilis,测定酶活及飞行时间质谱鉴定表明重组菌表达了具有活性的FAD-GDH。分别选取2种组成型启动子(PHpaⅡ-PamyQ’,PHpaⅡ-P43)和两种盐诱导型启动子(PHpaⅡ-PgsiB,PHpaⅡ-Popuaa)串联成双启动子分别表达FAD-GDH,含组成型双启动子PHpaⅡ-PamyQ’的重组B.subtilis,胞内表达量最高为2 497 U/L,PHpaⅡ-PamyQ’双启动子表达强度增加可能是由于转录强度增加。本实验中采用的其他双启动子表达FAD-GDH的活性低于单启动子,可能是启动子之间的排列顺序会影响启动之间的相互作用,靠近异源基因的启动子可能会更大程度地影响外源基因的表达[28]。在PHapII与gdh基因之间插入不同的启动子后,其RBS也被随之替换为新插入启动子的RBS,因此酶活性的变化也很有可能是由于翻译强度的变化引起的。
发酵过程中,发现葡萄糖和甘油会抑制串联启动子PamyQ’突变株的发酵产酶,存在碳代谢产物抑制效应。为了减少发酵中的碳代谢抑制,采取删去PamyQ’启动子中与碳代谢阻遏因子结合的cre位点,构建的含有缺失cre位点的双启动子PHpaⅡ-PamyQ2重组B.subtilis,使FAD-GDH酶活从2 497 U/L提高到3 626 U/L, 说明启动子PamyQ’中cre位点的删除有效地减少了碳源代谢产物对基因转录的抑制,促进外源基因表达。
参考文献
[1] TSUJIMURA S. From fundamentals to applications of bioelectrocatalysis: bioelectrocatalytic reactions of FAD-dependent glucose dehydrogenase and bilirubin oxidase[J]. Bioscience, Biotechnology, and Biochemistry, 2019, 83(1):39-48.
[2] ITO K, OKUDA S, JUNKO, et al. Designer fungus FAD glucose dehydrogenase capable of direct electron transfer[J]. Biosensors and Bioclectronics, 2019, 123: 114-123.
[3] GALANT A L, KAUFMAN R C, WILSON J D. Glucose: detection and analysis [J]. Food Chemistry, 2015, 188: 149-160.
[4] KOMORI H, INAKA K, FURUBAYASHI N, et al. Crystallographic analysis of FAD-dependent glucose dehydrogenase [J]. Acta Crystallographica Section F-Structural Biology Communications, 2015, 71(8): 1 017-1 019.
[5] YANG Yufeng, HUANG Lei, WANG Jufang, et al. Expression, characterization and mutagenesis of an FAD-dependent glucose dehydrogenase fromAspergillus terreus [J]. Enzyme and Microbial Technology, 2015, 68: 43-49.
[6] YOSHIDA H, SAKAI G, MORI K, et al. Structural analysis of fungus-derived FAD glucose dehydrogenase [J]. Scientific Reports, 2015, 5(1):13 498.
[7] 周利伟. 青霉来源葡萄糖脱氢酶的克隆、表达及其酶学性质研究[D]: 北京: 中国农业科学院, 2012.
[8] 田岛辽子, 一柳敦,市川惠一,等. 黄素结合型葡萄糖脱氢酶:中国,201080004947.X.[P].2010-04-19.
[9] YANG Yufeng, HUANG Lei, WANG Jufang, et al. Efficient expression, purification, and characterization of a novel FAD-dependent glucose dehydrogenase fromAspergillus terreus inPichia pastoris [J]. Journal of Microbiology and Biotechnology, 2014, 24(11): 1 516-1 524.
[10] YADA T, MIYAMOTO K. FAD-conjugated glucose dehydrogenase:United states, 9976125[P].2016-10-20.
[11] CHRISTOPH S, PETRA S, MIRIAM K, et al. Heterologous overexpression ofGlomerella cingulata FAD-dependent glucose dehydrogenase inEscherichia coli andPichia pastoris [J]. Microbial Cell Factories, 2011, 10:106.
[12] MURATA K, AKATSUKA W, SADAKANE T, et al. Glucose oxidation catalyzed by FAD-dependent glucose dehydrogenase within Os complex-tethered redox polymer hydrogel [J]. Electrochimica Acta, 2014, 136: 537-541.
[13] 余小霞,田健,刘晓青,等. 枯草芽孢杆菌表达系统及其启动子研究进展[J]. 生物技术通报, 2015, 31(2): 35-44.
[14] ZHANG K, SU L, DUAN X, et al. High-level extracellular protein production inBacillus subtilis using an optimized dual-promoter expression system [J]. Microbial Cell Factories, 2017, 16(1): 32-47.
[15] KANG H K, JANG J H, SHIM J H, et al. Efficient constitutive expression of thermostable 4-α-glucanotransferase inBacillus subtilis using dual promoters [J]. World Journal of Microbiology and Biotechnology, 2010, 26(10): 1 915-1 918.
[16] GUAN C, CUI W, CHENG J, et al. Construction of a highly active secretory expression system via an engineered dual promoter and a highly efficient signal peptide inBacillus subtilis [J]. New Biotechnology, 2016, 33(3): 372-379.
[17] BRUDER M, MOO Y M, CHUNG D A, et al. Elimination of carbon catabolite repression in clostridium acetobutylicum-a journey toward simultaneous use of xylose and glucose [J]. Applied Microbiology and Biotechnology, 2015, 99(18): 7 579-7 588.
[18] NICHOLSON W L, PARK Y K, HENKIN T M. Catabolite repression resistant mutations of theBacillus subtilis alpha amylase promoter affect transcription levels and are in an operator-like sequence [J]. Journal of Molecular Biology, 1987, 198(4): 609-618.
[19] NAGARAJAN D R, KRISHANA C. Use of a new catabolite repression resistant promoter isolated fromBacillus subtilis KCC103 for hyper-production of recombinant enzymes [J]. Protein Expression Purification, 2010, 70(1): 122-128.
[20] ZYPRIAN E, MATZURA H. Characterization of signals promoting gene expression on theStaphylococcus aureus plasmid pUB110 and development of a gram-positive expression vector system [J]. DNA, 1986, 5(3): 219-225.
[21] GUPTA M, RAO K K. Phosphorylation of DegU is essential for activation of amyE expression inBacillus subtilis [J]. Journal of Biosciences, 2014, 39(5): 747-752.
[22] WANG P Z, DOI R H. Overlapping promoters transcribed byBacillus subtilis sigma 55 and sigma 37 RNA polymerase holoenzymes during growth and stationary phases [J]. Biological Chemistry, 1984, 259(13): 8 619-8 625.
[23] KIM J H, HUANG B Y, ROH J. Comparison of PaprE, PamyE and P43 promoter strength for β-galactosidase and staphylokinase expression inBacillus subtilis [J]. Biotechnolgy and Bioprocess Engineering, 2008, 13(3): 313-318.
[24] MAUL B, VÖLKER U, RIETHDORF S, et al. σB-dependent regulation of gsiB in response to multiple stimuli inBacillus subtilis [J]. Molecular & General Genetics, 1995, 248(1): 114-120.
[25] PACCEZ J D, LUIZ W B, SBROGIO A M E, et al. Stable episomal expression system under control of a stress inducible promoter enhances the immunogenicity ofBacillus subtilis as a vector for antigen delivery [J]. Vaccine, 2006, 24(15): 2 935-2 943.
[26] PROMCHAI R, PROMDONKOY B, TANAPONGPIPAT S, et al. A novel salt-inducible vector for efficient expression and secretion of heterologous proteins inBacillus subtilis [J]. Journal of Biotechnology, 2016, 222: 86-93.
[27] 徐书景, 张跃灵,张妍,等. 改进重叠延伸PCR技术构建定点双突变[J]. 中国生物工程杂志, 2010, 30(10): 49-54.
[28] ZHANG K, SU L, DUAN X, et al. High-level extracellular protein production inBacillus subtilis using an optimized dual-promoter expression system [J]. Microbial Cell Factories, 2017, 16(1): 32-47.
Promoter tandem and transformation for FAD-conjugated glucose dehydrogenase expression in Bacillus subtilis
ZHANG Ling1, LIN Rong2, SONG Zukun1, WANG Nan1, YANG Hailin1*
1 (Key Laboratory of Industrial Biotechnology,Ministry of Education,Jiangnan University, Wuxi 214122, China) 2 (Suzhou Suncadia Biopharmaceuticals CO., Ltd, Suzhou 215000, China)
Abstract Glucose dehydrogenase (FAD-GDH, EC1.1.99.10),conjugated tightly with flavin adenine dinucleotide, is a novel diagnostic enzyme for the clinical detection of blood glucose. A protease-defective strainBacillus subtilis WB600 was used as a host to construct a shuttle plasmid pMA5-1 containing a single promoter PHpaII for expression of FAD-GDH gene (gdh) fromBurkholderia cepacia. The use of promoters in series and transformation strategies to investigate enzyme production. Four promoters (PamyQ', P43, PgsiB, Popuaa) were ligated with the promoter PHpaII on the plasmid,respectively. The results showed that the intracellular enzymatic activity of FAD-GDH was highest with PHpaII-PamyQ' tandem, which was 2 497 U/L. In order to reduce the inhibitory effect of glucose and glycerol on enzyme production during fermentation, on the basis of tandem combination, thecre sites binding to carbon metabolism regulatory proteins in PamyQ 'promoter were deleted, and the intracellular enzyme production level was increased to 3 626 U/L, indicating that the removal ofcre sites can reduce the inhibition of carbon metabolism products on promoter transcription. This study provides a reference for genetic modification and industrial production of a new diagnostic enzyme (FAD-GDH)
Key words flavin adenine dinucleotide (FAD); glucose dehydrogenase;Bacillus subtilis; tandem promoter;cre site
第一作者:硕士,助理研究员(杨海麟教授为通讯作者,E-mail:yanghailin@jiangnan.edu.cn)。
基金项目:江苏省产学研(BY2016022-40);国家轻工技术与工程一流学科自主课题资助(2018-23)
收稿日期:2018-09-04,改回日期:2019-01-18
DOI:10.13995/j.cnki.11-1802/ts.018685